
Ten artykuł szczegółowo wyjaśni, czym jest Zespół Gerstmanna-Sträusslera-Scheinkera (GSS) rzadka i poważna choroba neurologiczna. Poznasz jej przyczyny, objawy, metody diagnozy oraz dostępne formy wsparcia, co pozwoli na pełne zrozumienie tego skomplikowanego schorzenia.
Zespół Gerstmanna-Sträusslera-Scheinkera to rzadka, genetyczna choroba prionowa mózgu.
- GSS jest bardzo rzadką, dziedziczną chorobą neurozwyrodnieniową, zaliczaną do chorób prionowych.
- Wywoływana jest przez zmutowany gen PRNP, dziedziczony autosomalnie dominująco, co prowadzi do uszkodzeń mózgu przez patologiczne priony.
- Charakteryzuje się postępującą ataksją móżdżkową (problemy z koordynacją) i późno pojawiającą się demencją.
- Objawy pojawiają się zazwyczaj między 35. a 55. rokiem życia, a przebieg choroby trwa średnio od 2 do 10 lat.
- Obecnie nie ma lekarstwa; leczenie jest wyłącznie objawowe i paliatywne, a rokowania są niepomyślne.
- Akronim "GSA" w kontekście medycznym odnosi się do tej choroby, a nie do zjawiska psychologicznego "Genetic Sexual Attraction".
Czym jest syndrom GSA? Wyjaśniamy zagadkę rzadkiej choroby prionowej
W świecie medycyny istnieje wiele chorób, które choć rzadkie, niosą ze sobą ogromne wyzwania diagnostyczne i terapeutyczne. Jedną z nich jest Zespół Gerstmanna-Sträusslera-Scheinkera (GSS), niezwykle rzadka, genetycznie uwarunkowana choroba neurozwyrodnieniowa. To schorzenie, choć mniej znane niż inne choroby neurologiczne, zasługuje na naszą uwagę ze względu na swój postępujący i wyniszczający charakter. GSS należy do grupy pasażowalnych encefalopatii gąbczastych, co oznacza, że jest to choroba prionowa, blisko spokrewniona z takimi schorzeniami jak choroba Creutzfeldta-Jakoba.
Zespół Gerstmanna-Sträusslera-Scheinkera: Pełna nazwa i jej znaczenie
Pełna nazwa choroby Zespół Gerstmanna-Sträusslera-Scheinkera upamiętnia trzech neurologów: Josefa Gerstmanna, Ernsta Sträusslera i Ilję Scheinkera, którzy jako pierwsi opisali to schorzenie. Klasyfikacja GSS jako choroby neurozwyrodnieniowej jest kluczowa. Oznacza to, że dochodzi w niej do stopniowego i nieodwracalnego uszkodzenia komórek nerwowych w mózgu. W konsekwencji prowadzi to do postępującej utraty funkcji neurologicznych, co jest niezwykle poważne dla całego układu nerwowego i życia pacjenta.
Dlaczego to choroba prionowa i co to oznacza dla mózgu?
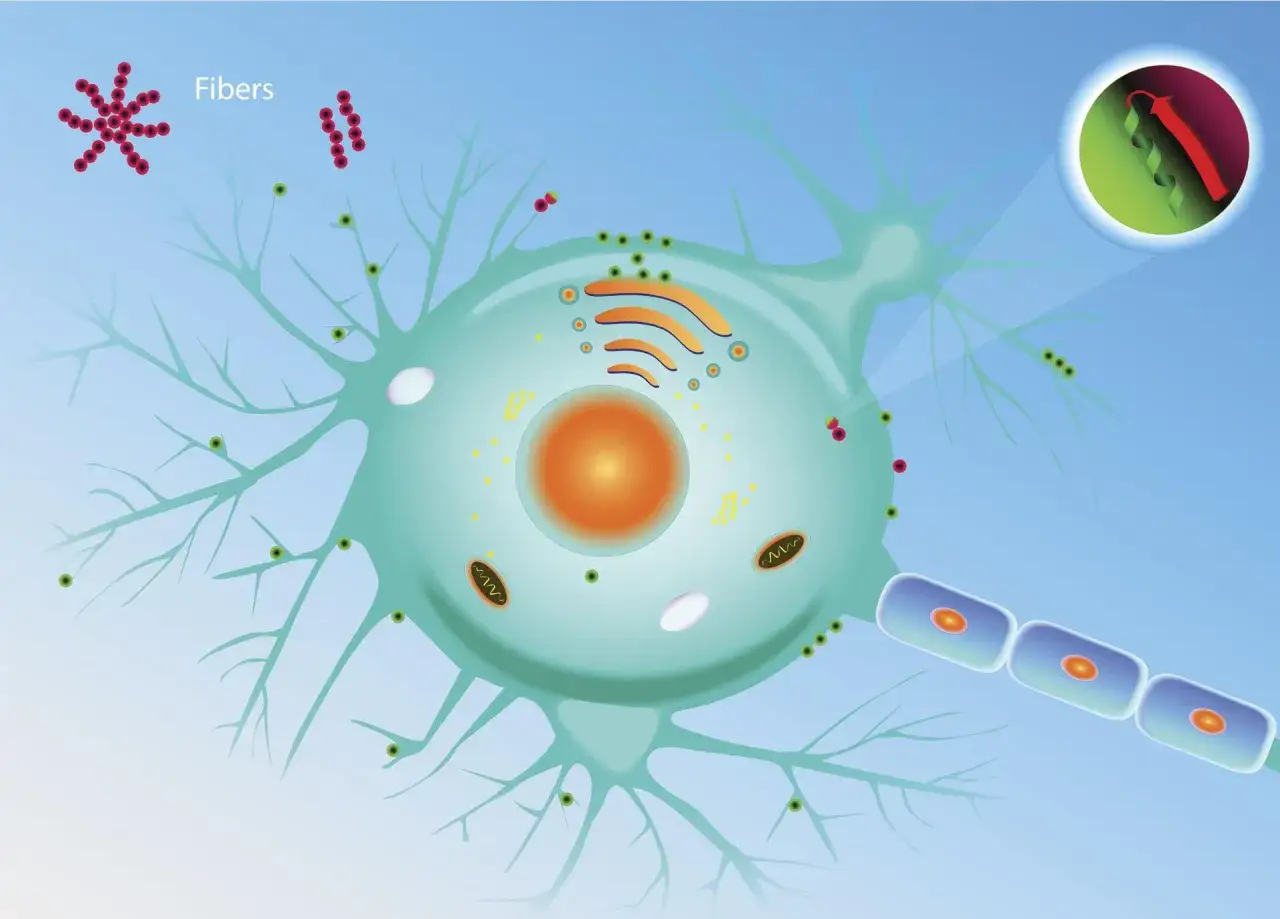
Zrozumienie, czym są priony, jest fundamentalne dla pojęcia GSS. Priony to białkowe cząsteczki zakaźne, które w swojej prawidłowej formie występują naturalnie w organizmie, szczególnie w mózgu. Jednak w przypadku chorób prionowych, takich jak GSS, dochodzi do ich nieprawidłowego zwijania się i tworzenia patologicznych form. Te zmienione priony są niezwykle odporne na degradację i mają zdolność do "zarażania" innych, prawidłowo zwiniętych białek, zmieniając ich strukturę. W efekcie, w mózgu gromadzą się agregaty patologicznych prionów, co prowadzi do uszkodzenia i śmierci neuronów. Mózg staje się dosłownie "gąbczasty" stąd nazwa encefalopatia gąbczasta co objawia się licznymi wakuolami (pustymi przestrzeniami) i ubytkami tkanki nerwowej. To właśnie priony są kluczowym elementem patogenezy GSS, odpowiedzialnym za jego destrukcyjny wpływ na mózg.
GSA jako choroba neurologiczna a GSA jako zjawisko psychologiczne: Kluczowe rozróżnienie
W kontekście medycznym akronim "GSA" odnosi się do Zespołu Gerstmanna-Sträusslera-Scheinkera. Należy jednak zwrócić uwagę, że w przestrzeni internetowej i kulturowej akronim "GSA" może być również używany w odniesieniu do "Genetic Sexual Attraction" (Genetyczne Przyciąganie Seksualne), które jest zjawiskiem psychologicznym, opisującym silne uczucia atrakcji między bliskimi krewnymi, którzy spotykają się po raz pierwszy jako dorośli. Jest to niezwykle ważne rozróżnienie, ponieważ te dwa pojęcia nie mają ze sobą nic wspólnego. W tym artykule koncentrujemy się wyłącznie na aspekcie medycznym, czyli na Zespole Gerstmanna-Sträusslera-Scheinkera, aby uniknąć jakichkolwiek nieporozumień i dostarczyć rzetelnych informacji na temat tej rzadkiej choroby neurologicznej.

Jakie sygnały alarmowe wysyła organizm? Kluczowe objawy syndromu GSA
Zrozumienie objawów Zespołu Gerstmanna-Sträusslera-Scheinkera jest kluczowe, choć diagnoza jest trudna ze względu na rzadkość choroby. Pierwsze sygnały alarmowe pojawiają się zazwyczaj między 35. a 55. rokiem życia, co jest stosunkowo wcześnie w porównaniu do innych chorób neurozwyrodnieniowych. Choroba ma charakter postępujący, a jej przebieg trwa średnio od 2 do 10 lat. W tym czasie objawy stopniowo się nasilają, prowadząc do znacznego pogorszenia stanu pacjenta. Przyjrzyjmy się głównym symptomom, które powinny wzbudzić niepokój.
Ataksja móżdżkowa: Kiedy problemy z równowagą i chodem stają się niepokojące
Jednym z najbardziej charakterystycznych i często pierwszych objawów GSS jest postępująca ataksja móżdżkowa. Móżdżek jest częścią mózgu odpowiedzialną za koordynację ruchów, równowagę i precyzję. Kiedy zostaje uszkodzony przez patologiczne priony, pacjenci zaczynają doświadczać coraz większych trudności w utrzymaniu równowagi, ich chód staje się niepewny, szeroki i chwiejny, przypominający chód osoby pod wpływem alkoholu. Częste upadki stają się codziennością, a wykonywanie prostych czynności, takich jak sięganie po przedmioty czy pisanie, staje się coraz trudniejsze. To znacząco wpływa na samodzielność i jakość życia, sprawiając, że codzienne funkcjonowanie staje się prawdziwym wyzwaniem.
Demencja i zaburzenia poznawcze: Jak choroba wpływa na umysł?
W przeciwieństwie do niektórych innych chorób prionowych, w GSS otępienie (demencja) pojawia się stosunkowo późno w przebiegu choroby. Początkowo pacjenci mogą doświadczać subtelnych problemów z pamięcią, koncentracją czy trudnościami w znajdowaniu odpowiednich słów. W miarę postępu choroby, zaburzenia poznawcze nasilają się, prowadząc do znacznego upośledzenia funkcji intelektualnych. Pacjenci mogą mieć problemy z orientacją w czasie i przestrzeni, podejmowaniem decyzji, rozwiązywaniem problemów, a także z rozpoznawaniem bliskich osób. W zaawansowanych stadiach demencja staje się głęboka, uniemożliwiając jakąkolwiek samodzielność.
Od zaburzeń mowy po parkinsonizm: Inne istotne objawy neurologiczne
Oprócz ataksji i demencji, Zespół Gerstmanna-Sträusslera-Scheinkera może manifestować się szeregiem innych objawów neurologicznych, które dodatkowo pogarszają stan pacjenta. Należą do nich:
- Dyzartria: Zaburzenia mowy, polegające na trudnościach w artykulacji słów, co sprawia, że mowa staje się niewyraźna i trudna do zrozumienia.
- Oczopląs: Mimowolne, rytmiczne ruchy gałek ocznych, które mogą prowadzić do zaburzeń widzenia.
- Zaburzenia widzenia: Poza oczopląsem, mogą występować inne problemy z widzeniem, choć rzadziej niż w innych chorobach prionowych.
- Parkinsonizm: Objawy przypominające chorobę Parkinsona, takie jak spowolnienie ruchowe (bradykinezja), sztywność mięśniowa i drżenie spoczynkowe.
- Mioklonie: Krótkie, nagłe, mimowolne zrywy mięśniowe, które mogą występować w różnych częściach ciała.
- Wzmożone napięcie mięśniowe: Spastyczność, czyli zwiększona sztywność mięśni, utrudniająca ruchy.
Jak choroba postępuje? Typowy przebieg od pierwszych symptomów
Typowy przebieg GSS charakteryzuje się niestety stałym i nieubłaganym pogarszaniem się stanu zdrowia. Zaczyna się od subtelnych problemów z koordynacją lub równowagą, które często są początkowo bagatelizowane lub przypisywane innym przyczynom. Z czasem objawy te stają się coraz bardziej widoczne i uciążliwe. Problemy z chodem nasilają się, pacjent wymaga wsparcia, a następnie wózka inwalidzkiego. Pojawiają się zaburzenia mowy, trudności w połykaniu, a także coraz głębsze deficyty poznawcze. W zaawansowanych stadiach pacjenci często są przykuci do łóżka, wymagają pełnej opieki i stają się całkowicie zależni od innych. To postępujący i wyniszczający charakter GSS sprawia, że jest to choroba tak tragiczna w skutkach, zarówno dla pacjentów, jak i ich bliskich.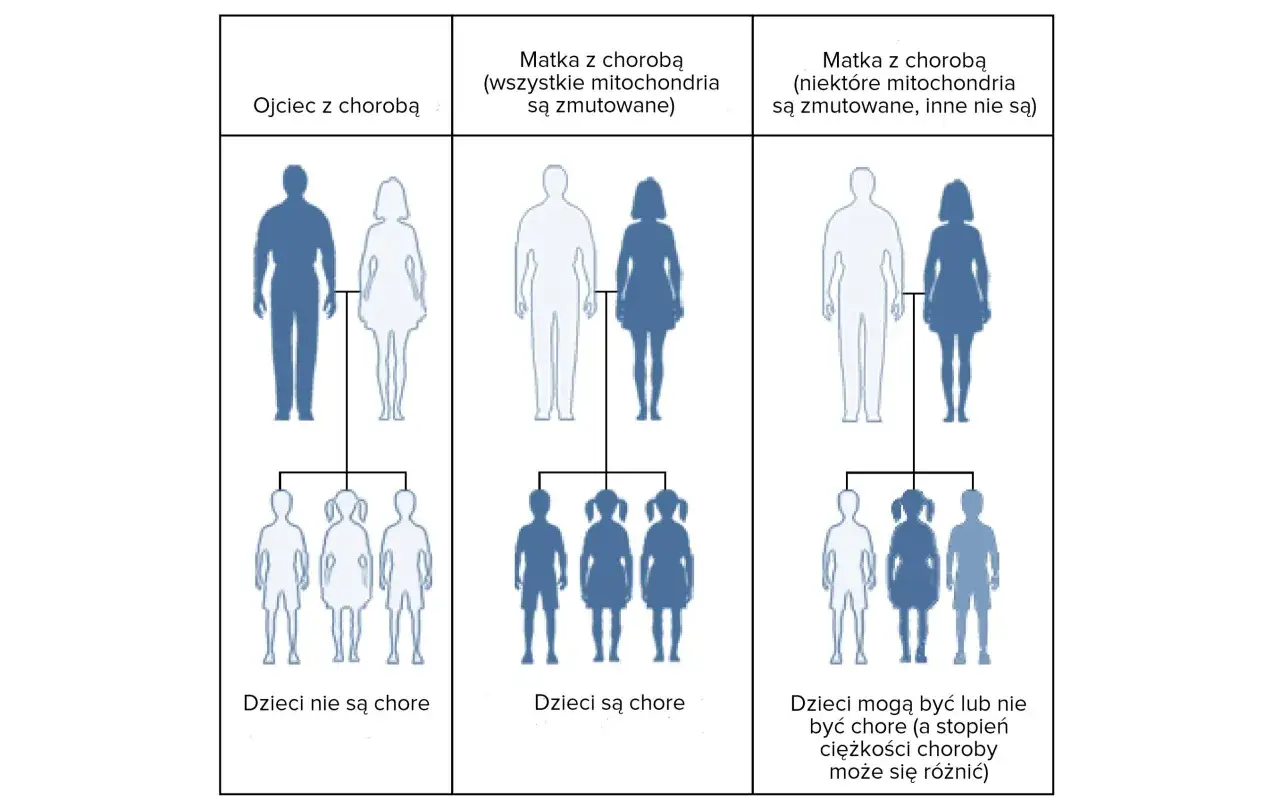
Skąd bierze się syndrom GSA? Przyczyny genetyczne w centrum uwagi
Zrozumienie etiologii Zespołu Gerstmanna-Sträusslera-Scheinkera jest kluczowe, a w jego przypadku przyczyną leżącą u podstaw jest genetyka. GSS nie jest chorobą nabytą w wyniku infekcji czy czynników środowiskowych, lecz jest ściśle związana z naszym materiałem genetycznym. To sprawia, że ma ona charakter rodzinny, co ma ogromne znaczenie dla planowania rodziny i doradztwa genetycznego.
Rola zmutowanego genu PRNP: Genetyczny korzeń choroby
Główną i bezpośrednią przyczyną Zespołu Gerstmanna-Sträusslera-Scheinkera jest obecność germinalnej mutacji w genie PRNP. Ten gen koduje białko prionowe (PrP), które, jak wspomniałem wcześniej, w swojej prawidłowej formie jest nieszkodliwe i pełni ważne funkcje w organizmie. Jednak mutacja w genie PRNP prowadzi do tego, że białko prionowe jest produkowane w nieprawidłowej, patologicznej formie (PrPSc). To właśnie te zmienione priony są odpowiedzialne za rozwój choroby. Mają one tendencję do agregacji i tworzenia płytek amyloidowych w mózgu, co prowadzi do uszkodzenia neuronów i charakterystycznych zmian gąbczastych. Różne mutacje w genie PRNP mogą prowadzić do różnych fenotypów chorób prionowych, a w przypadku GSS najczęściej są to mutacje w kodonie 102 (P102L), 105, 117, 145, 198, 217.
Dziedziczenie autosomalne dominujące: Jakie jest ryzyko przekazania choroby dzieciom?
Zespół Gerstmanna-Sträusslera-Scheinkera dziedziczy się w sposób autosomalny dominujący. Co to oznacza w praktyce? Jeśli jeden z rodziców jest nosicielem zmutowanego genu PRNP i jest chory na GSS, istnieje 50% ryzyka, że każde z jego dzieci również odziedziczy ten zmutowany gen i rozwinie chorobę. Nie ma znaczenia płeć dziecka ani płeć chorego rodzica. Każde potomstwo ma równe szanse na odziedziczenie wadliwego genu. Jest to niezwykle ważna informacja dla rodzin dotkniętych GSS, ponieważ pozwala na świadome podejmowanie decyzji dotyczących planowania rodziny i ewentualne skorzystanie z poradnictwa genetycznego.
Czy choroba może pojawić się spontanicznie? Mutacje "de novo"
Chociaż GSS jest chorobą genetyczną i zazwyczaj ma charakter dziedziczny, istnieją również bardzo rzadkie przypadki, w których choroba pojawia się bez wcześniejszej historii rodzinnej. Mówimy wtedy o mutacjach "de novo". Oznacza to, że mutacja w genie PRNP powstaje spontanicznie u danej osoby, bez odziedziczenia jej od żadnego z rodziców. Taka sytuacja jest rzadka, ale możliwa. W przypadku mutacji "de novo" dana osoba jest pierwszą w rodzinie, u której występuje choroba, jednak jej potomstwo będzie już miało 50% ryzyko odziedziczenia zmutowanego genu, zgodnie z zasadami dziedziczenia autosomalnego dominującego.
Jak lekarze stawiają diagnozę? Proces rozpoznawania syndromu GSA
Postawienie diagnozy Zespołu Gerstmanna-Sträusslera-Scheinkera jest procesem złożonym, wymagającym połączenia obserwacji klinicznych, szczegółowego wywiadu oraz zaawansowanych badań laboratoryjnych i genetycznych. Ze względu na rzadkość choroby i niespecyficzne objawy początkowe, diagnoza może być opóźniona. Moim zdaniem, kluczowe jest podejście interdyscyplinarne i świadomość istnienia tego schorzenia wśród neurologów.
Od wywiadu rodzinnego do badania neurologicznego: Pierwsze kroki w diagnostyce
Proces diagnostyczny zazwyczaj rozpoczyna się od szczegółowego wywiadu rodzinnego. Ze względu na dziedziczny charakter GSS, informacja o występowaniu podobnych objawów neurologicznych lub zdiagnozowanej chorobie w rodzinie jest niezwykle cenna. Następnie lekarz przeprowadza dokładne badanie neurologiczne, oceniając objawy takie jak ataksja móżdżkowa, zaburzenia mowy, problemy z równowagą, obecność mioklonii czy cech parkinsonizmu. Obraz kliniczny, czyli zestaw obserwowanych objawów, jest podstawą do podejrzewania GSS, zwłaszcza gdy objawy postępują i nie pasują do innych, częstszych schorzeń.
Badania genetyczne: Ostateczne potwierdzenie obecności mutacji
W przypadku podejrzenia Zespołu Gerstmanna-Sträusslera-Scheinkera, badanie genetyczne w kierunku mutacji w genie PRNP jest kluczowe i stanowi ostateczne potwierdzenie diagnozy. Analiza DNA pacjenta pozwala na wykrycie specyficznych mutacji, które są odpowiedzialne za rozwój choroby. Jest to metoda bardzo precyzyjna i niezbędna do jednoznacznego rozpoznania GSS, odróżniając ją od innych chorób neurozwyrodnieniowych. Badanie genetyczne może być również rozważane u członków rodziny pacjenta, którzy są w grupie ryzyka, w ramach poradnictwa genetycznego.
Rola badań obrazowych (MRI, TK) w wykluczaniu innych schorzeń
Badania obrazowe mózgu, takie jak rezonans magnetyczny (MRI) i tomografia komputerowa (TK), odgrywają ważną rolę w diagnostyce różnicowej GSS. Nie są one narzędziami do bezpośredniej diagnozy GSS, ponieważ zmiany charakterystyczne dla chorób prionowych mogą być subtelne lub pojawiać się dopiero w zaawansowanych stadiach. Ich głównym celem jest wykluczenie innych schorzeń neurologicznych, które mogą dawać podobne objawy, takich jak guzy mózgu, udary, stwardnienie rozsiane czy inne formy demencji. MRI może wykazać zaniki mózgu, szczególnie w obrębie móżdżku, ale te zmiany nie są specyficzne dla GSS. Warto również wspomnieć, że ostateczne potwierdzenie obecności charakterystycznych dla GSS złogów amyloidu, które są zbudowane z patologicznych prionów, możliwe jest jedynie w badaniu neuropatologicznym mózgu, które przeprowadza się pośmiertnie. To badanie dostarcza najbardziej precyzyjnych informacji o zmianach mikroskopowych w tkance mózgowej.
Czy syndrom GSA można leczyć? Obecne możliwości terapeutyczne i rokowania
Niestety, w przypadku Zespołu Gerstmanna-Sträusslera-Scheinkera, muszę przekazać informację, która jest trudna do przyjęcia zarówno dla pacjentów, jak i ich rodzin. Obecny stan wiedzy medycznej nie pozwala na wyleczenie tej choroby. Jest to aspekt, który budzi wiele frustracji i smutku, ale jest to rzeczywistość, z którą musimy się zmierzyć. Skupmy się na tym, co jest możliwe do zrobienia w obliczu tej diagnozy.
Dlaczego nie ma lekarstwa? Wyzwania w leczeniu chorób prionowych
Zespół Gerstmanna-Sträusslera-Scheinkera, podobnie jak inne choroby prionowe, jest obecnie chorobą nieuleczalną. Brak skutecznego lekarstwa wynika ze specyfiki patogenezy tych schorzeń. Patologiczne priony są białkami własnego organizmu, co utrudnia rozwój leków, które selektywnie niszczyłyby tylko ich zmienioną formę, nie uszkadzając jednocześnie prawidłowych białek. Ponadto, priony są niezwykle odporne na standardowe metody dezynfekcji i leczenia, a ich akumulacja w mózgu prowadzi do nieodwracalnych uszkodzeń neuronów. Wyzwania w leczeniu chorób prionowych są ogromne, a badania nad nowymi terapiami są intensywnie prowadzone, choć na razie bez przełomowych sukcesów.
Leczenie objawowe: Jak poprawić jakość życia pacjentów?
Mimo braku leczenia przyczynowego, opieka nad pacjentami z GSS koncentruje się na leczeniu objawowym i paliatywnym. Celem jest maksymalne poprawienie jakości życia pacjenta, łagodzenie cierpienia i zapewnienie komfortu w miarę postępu choroby. Przykłady działań w ramach leczenia objawowego obejmują:
- Fizjoterapia: Pomaga w utrzymaniu sprawności ruchowej tak długo, jak to możliwe, zmniejsza spastyczność i zapobiega przykurczom.
- Farmakoterapia objawowa: Leki mogą być stosowane do łagodzenia poszczególnych objawów, takich jak mioklonie (leki przeciwdrgawkowe), sztywność mięśniowa (leki rozluźniające mięśnie) czy zaburzenia snu.
- Wsparcie żywieniowe: W miarę postępu problemów z połykaniem, konieczne może być zastosowanie specjalnych diet lub żywienia dojelitowego.
- Opieka psychologiczna: Zarówno dla pacjenta, jak i rodziny, aby pomóc im radzić sobie z diagnozą i postępem choroby.
Choć te interwencje nie zatrzymują postępu GSS, są niezwykle ważne dla godności i komfortu pacjenta.
Rokowania i średni czas przeżycia: Co mówią dane medyczne?
Rokowania dla pacjentów z Zespołem Gerstmanna-Sträusslera-Scheinkera są niestety niepomyślne. Choroba jest postępująca i zawsze prowadzi do śmierci. Średni czas przeżycia od wystąpienia pierwszych objawów wynosi zazwyczaj od 2 do 10 lat, choć w zależności od konkretnej mutacji i indywidualnego przebiegu choroby, może być on nieco krótszy lub dłuższy. Należy pamiętać, że jest to choroba wyniszczająca, która stopniowo pozbawia pacjenta samodzielności i wszystkich funkcji życiowych. Ten aspekt jest szczególnie trudny dla rodzin, które muszą być przygotowane na długotrwałą i intensywną opiekę.
Życie z diagnozą GSS: Wsparcie dla pacjentów i ich rodzin
Diagnoza Zespołu Gerstmanna-Sträusslera-Scheinkera to wyrok, który zmienia życie pacjenta i jego bliskich w sposób fundamentalny. Ze względu na rzadkość i powagę choroby, kluczowe staje się zapewnienie kompleksowego wsparcia, które obejmuje zarówno aspekty medyczne, jak i psychologiczne. Moim zdaniem, nikt nie powinien być sam w obliczu tak trudnej sytuacji.
Rola opieki paliatywnej i multidyscyplinarnego zespołu specjalistów
W przypadku GSS, gdzie leczenie przyczynowe jest niemożliwe, opieka paliatywna odgrywa centralną rolę. Jej celem jest zapewnienie jak największego komfortu, łagodzenie bólu i innych objawów, a także dbałość o godność pacjenta na każdym etapie choroby. Opieka ta jest zazwyczaj świadczona przez multidyscyplinarny zespół specjalistów, który może obejmować:
- Neurologów: Monitorujących postęp choroby i dostosowujących leczenie objawowe.
- Fizjoterapeutów: Pomagających w utrzymaniu sprawności ruchowej i zapobieganiu powikłaniom.
- Logopedów: Wspierających pacjentów z dyzartrią i problemami z połykaniem.
- Dietetyków: Dbających o odpowiednie odżywienie.
- Psychologów/psychoterapeutów: Oferujących wsparcie emocjonalne.
- Pielęgniarki paliatywne: Zapewniające codzienną opiekę i wsparcie.
Taki zespół pracuje wspólnie, aby sprostać złożonym potrzebom pacjenta i jego rodziny.
Wsparcie psychologiczne dla chorych i ich bliskich w obliczu rzadkiej choroby
Diagnoza GSS jest traumatycznym doświadczeniem. Pacjenci muszą zmierzyć się z perspektywą postępującej utraty funkcji i nieuchronnej śmierci, a ich bliscy z wizją utraty ukochanej osoby i koniecznością sprawowania coraz bardziej wymagającej opieki. Wsparcie psychologiczne jest w tej sytuacji absolutnie kluczowe. Psychologowie i terapeuci mogą pomóc w radzeniu sobie z żałobą antycypacyjną, lękiem, depresją i poczuciem bezradności. Ważne jest, aby zarówno pacjenci, jak i ich rodziny mieli dostęp do profesjonalnej pomocy, która pozwoli im przetworzyć emocje i znaleźć strategie radzenia sobie z tą niezwykle trudną sytuacją. Grupy wsparcia dla rodzin pacjentów z chorobami rzadkimi mogą również okazać się nieocenionym źródłem pocieszenia i praktycznych porad.
Przeczytaj również: ME/CFS: Objawy, PEM, diagnoza. Czy to już choroba?
Gdzie szukać wiarygodnych informacji i pomocy w Polsce?
W obliczu tak rzadkiej i skomplikowanej choroby, dostęp do wiarygodnych informacji i wsparcia jest na wagę złota. W Polsce pacjenci i ich rodziny mogą szukać pomocy w następujących miejscach:
- Specjalistyczne ośrodki neurologiczne: Uniwersyteckie kliniki neurologiczne często posiadają największe doświadczenie w diagnozowaniu i prowadzeniu pacjentów z rzadkimi chorobami neurozwyrodnieniowymi.
- Stowarzyszenia pacjentów z chorobami rzadkimi: Takie organizacje mogą oferować wsparcie informacyjne, emocjonalne, a czasem także dostęp do grup wsparcia. Warto poszukać stowarzyszeń zajmujących się chorobami neurozwyrodnieniowymi lub chorobami prionowymi.
- Fundacje medyczne: Niektóre fundacje wspierają badania nad chorobami rzadkimi lub oferują pomoc pacjentom i ich rodzinom.
- Poradnie genetyczne: W przypadku chorób dziedzicznych, poradnictwo genetyczne jest niezbędne do zrozumienia ryzyka i możliwości.
- Internetowe bazy danych i fora: Zawsze należy podchodzić do nich z ostrożnością, ale mogą stanowić źródło wymiany doświadczeń, choć zawsze należy weryfikować informacje u źródeł medycznych.
Pamiętajmy, że choć GSS jest chorobą rzadką i nieuleczalną, odpowiednie wsparcie i opieka mogą znacząco poprawić jakość życia pacjentów i pomóc ich rodzinom w przejściu przez ten trudny czas.